Notes
L’accès à cette page nécessite une autorisation. Vous pouvez essayer de vous connecter ou de modifier des répertoires.
L’accès à cette page nécessite une autorisation. Vous pouvez essayer de modifier des répertoires.
Le but de la validation est d’assurer la cohérence d’un processus ou système et qu’il soit documenté. La validation du système est une exigence des organismes de réglementation. Pour les organisations des sciences de la vie, par exemple, les organismes de réglementation comprennent la Food and Drug Administration (FDA) des États-Unis.
La FDA définit la validation comme suit :
Confirmation par examen et fourniture de preuves objectives que les exigences particulières pour une utilisation spécifique prévue peuvent être satisfaites de manière cohérente.
L’Organisation mondiale de la santé (OMS) définit la validation comme suit dans ses directives sur les exigences relatives aux bonnes pratiques de fabrication (BPF) :
Établissement de preuves documentaires qui fournissent un degré élevé d’assurance qu’un processus planifié sera uniformément conforme aux résultats spécifiés attendus.
Ces définitions ont en commun les éléments suivants, conformément aux résultats attendus :
- Génération de preuves
- Conformité avec les réglementations
- Exécution des obligations
La validation des systèmes informatisés est un processus documenté visant à garantir que le système fait exactement ce pour quoi il a été conçu, de manière cohérente et reproductible. La validation garantit l’intégrité et la sécurité du traitement des données, la qualité des produits et la conformité aux réglementations applicables aux bonnes pratiques {industry} (GxP).
Le processus de validation d’un système informatisé est décrite dans les procédures opérationnelles standard (SOP) et les directives créées et définies par l’industrie réglementée, comme les organisations des sciences de la vie. Pour la validation de systèmes informatisés, il est utile de considérer la mise en œuvre du processus comme un projet, tel que décrit dans l’International Society for Pharmaceutical Engineering (ISPE), Good Automated Manufacturing Practice (GAMP) 5 : approche basée sur les risques pour des systèmes informatisés conformes aux BxP.
Avant de commencer le projet de mise en œuvre, un plan global pour la nouvelle solution doit être en place. Démarrez ensuite le projet en réalisant les phases suivantes :
- Planification : au cours de cette phase, les exigences et les spécifications doivent être suffisamment claires pour une évaluation initiale des risques et finalement pour une définition correcte des tests de vérification (protocoles). Au cours de cette phase, vous livrez le document de plan de validation qui définit l’ensemble de la stratégie de validation et de tous les livrables. La stratégie doit être conforme au système et aux stratégies de gestion de la qualité (QMS).
- Spécification, configuration et codage : au cours de cette phase, toutes les spécifications de conception sont établies avec le niveau de détail requis par le type de système et son utilisation. Les développeurs choisissent et utilisent les méthodes et objets de développement les plus appropriés aux exigences de codage et de configuration et basés sur les spécifications approuvées. Toutes ces activités sont réalisées dans l’environnement de développement. Pendant cette phase, les tests sont davantage axés sur la vérification des unités ou des fonctionnalités du point de vue du développeur. Parmi les exemples d’activités de test, citons les tests unitaires, les tests statistiques du code et les tests d’intégration. Les outils peuvent automatiser ces activités de test.
- Test : cette phase confirme que les spécifications ont été respectées grâce aux inspections et aux tests du système. Les activités de test sont effectuées dans un environnement de test préparé et adapté. L’environnement de test doit ressembler à l’environnement de production pour garantir que les conditions sont identiques et que vous n’avez pas besoin de répéter les tests dans l’environnement de production. Le risque doit déterminer la portée de l’effort de test. L’analyse des risques peut vous aider à comprendre les dangers potentiels susceptibles d’avoir un impact sur la qualité du produit, la sécurité des patients ou l’intégrité des données. Ces dangers potentiels doivent être atténués par les contrôles mise en place et la preuve des tests. S’il existe un risque élevé quelque part, veillez à disposer de scénarios de test appropriés pour prouver que la conception de la solution est sans défaillance potentielle.
- Rapport et publication : au cours de cette phase, le système doit être acceptable pour une utilisation dans l’environnement de production selon un processus documenté et contrôlé. À la clôture du projet, une réalisation de la validation du système doit être préparée pour résumer les activités mises en œuvre et tout écart éventuel par rapport au plan de validation. La validation du système doit être achevée avant la mise en service du système.
L’illustration suivante montre le modèle en V pris en charge dans la 2e édition du GAMP 5. Elle offre une bonne vision globale des phases du projet.
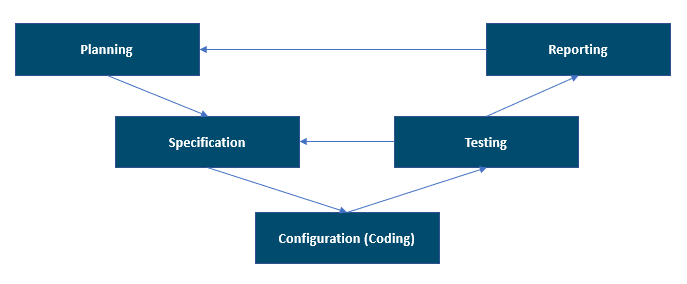
Le modèle en V peut être considéré non seulement comme les activités de développement et de test du système, mais aussi leur séquence, leurs interrelations et le processus de validation des livrables applicables au système informatisé validé. Vous devez conserver et maintenir les interrelations entre les exigences, les spécifications et les tests. Cette interrelation est documentée dans la matrice de traçabilité utilisée dans les zones réglementées.
La matrice de traçabilité garantit que :
- Les obligations sont respectées par la conception de la solution. En d’autres termes, chaque obligation est liée aux fonctions, commandes, configurations ou éléments de conception.
- Les obligations sont testées ou vérifiées pour faire la démonstration que la conception de la solution répond aux exigences, selon le cas.
La Matrice de traçabilité présent les avantages suivants :
- Elle prend en charge la revue de conception.
- Elle permet de définir la portée des tests de régression.
- Elle fournit un soutien lors des activités d’inspection ou d’audit.
- Elle fournit un soutien pour les changements potentiels.
Qualification de plate-forme
Les industries réglementées doivent qualifier la Microsoft Power Platform en tant qu’infrastructure avant la mise en œuvre de Guides. La qualification de la plate-forme requière les tâches suivantes au minimum :
Évaluation initiale des risques (évaluation de l’applicabilité GxP)
Évaluation du fournisseur (audit virtuel, physique ou postal d’un fournisseur)
Plan de qualification
Spécification technique de conception de la plate-forme
Évaluation des risques, par exemple l’évaluation du risque de créer la mauvaise version d’un guide mise à la disposition des opérateurs
Tests :
- Tests d’installation, par exemple tester que les environnements sont correctement installés
- Tests opérationnels, par exemple tester que les bons utilisateurs disposent des bons accès
Rapport de synthèse des qualifications
Plate-forme ou manuel opérationnel, support de formation
Validations des applications
Les applications (telles que Guides et Power Apps) qui prennent en charge les processus métier dans les secteurs réglementés doivent être validées. Par conséquent, votre organisation doit effectuer les tâches suivantes :
Évaluation initiale des risques
Plan de validation
Exigences utilisateur
Évaluation des risques
Spécification fonctionnelle ou spécification technique de configuration de l’application
Tests (qualification de l’installation [IQ], qualification opérationnelle [OQ] et test d’acceptation par l’utilisateur [UAT]) :
- Tests opérationnels, par exemple la vérification d’une fonction
- UAT
Matrice de traçabilité
Rapport de synthèse de la validation
Application ou manuel opérationnel, support de formation